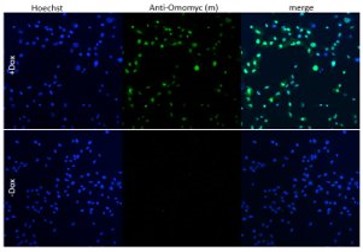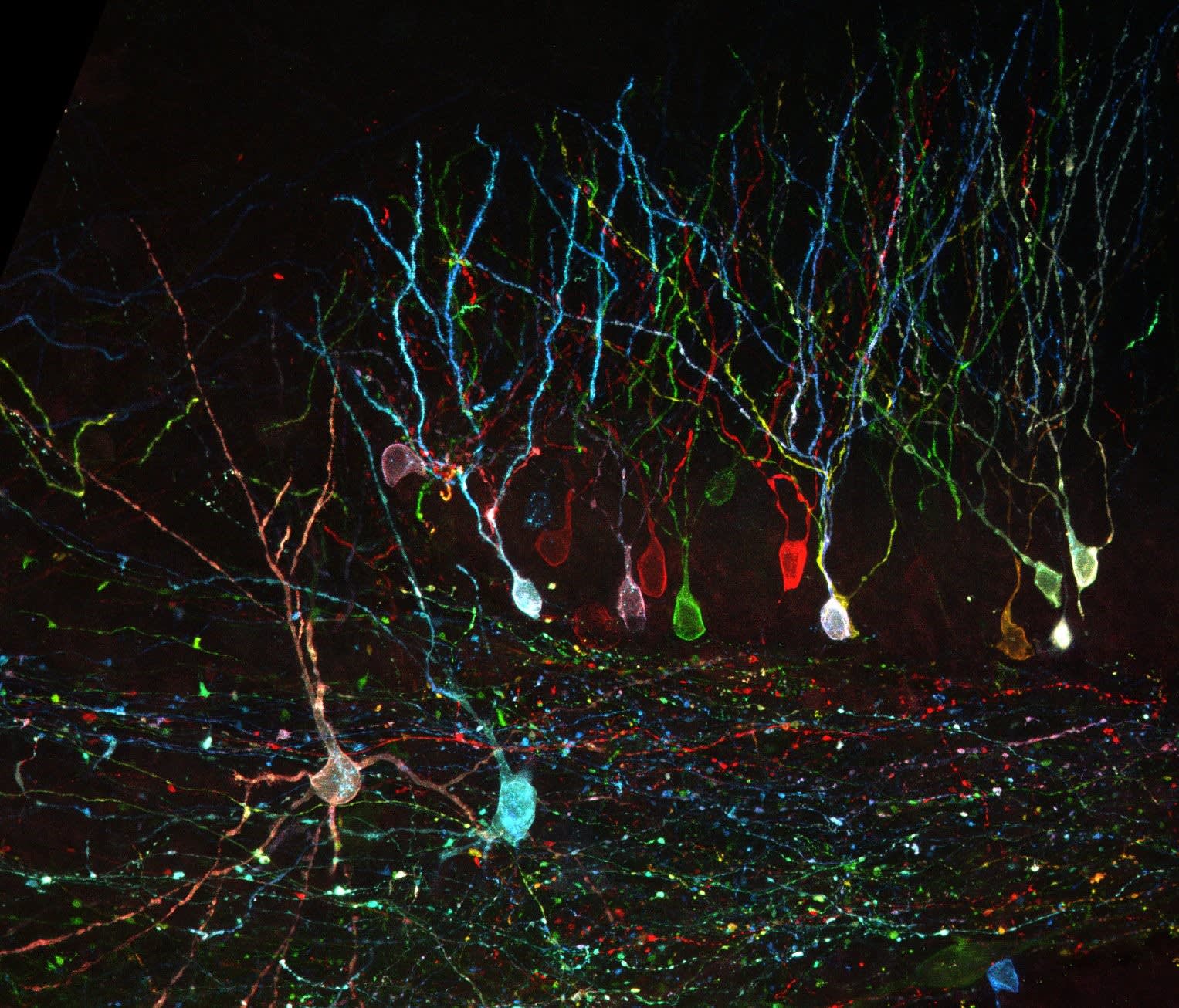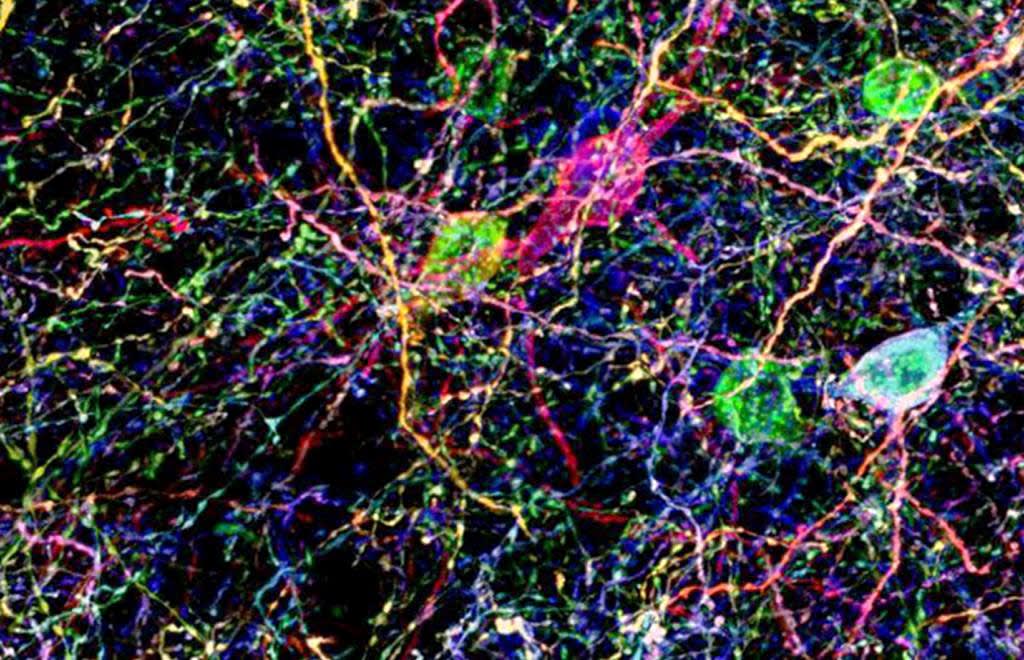
Introduction
For over a century, researchers have been focused on understanding how tumours evade the immune response and thrive. This research has identified key interactions between immune cells and cancer cells, leading to the development of many novel oncology treatments and improved prognosis for many patients.
Image: Light micrograph of a section through a lymph node affected by Hodgkin’s lymphoma, a cancer of lymphoma white blood cells. Stock image.
Tumour immunology encompasses the study of interactions between tumours and the immune system, focusing on understanding how tumours can evade immune responses and thrive. The origin of tumour immunology can be attributed to William Coley, who created the Coley toxin – a heat-inactivated streptococcal cocktail, used to treat sarcomas in the 1890s successfully (1). This treatment was created to stimulate what we now understand to be the immune system to fight cancer. The Coley toxin fell out of favour with the discovery of other oncological treatments such as chemotherapy and radiology (1). This divide in approach between the fields of cancer and immunology continued until around the 90s when fundamental discoveries around inhibiting immune checkpoints were made, ultimately leading to a noble prize for Allison and Honjo in 2018 for their work on cancer immunology (2).
Despite significant advances in the field and the successful implementation of therapeutics that target the immune response, the battle within tumour immunology continues with challenges, such as immunotherapy resistance and efficacy. Here we discuss some of the current barriers within tumour immunology, such as immunotherapy efficacy, the tumour microenvironment (TME), as well as potential avenues being explored to overcome this resistance.
A challenging environment: Why is the tumour microenvironment such a formidable opponent to immunotherapies?
The TME is a complex and dense battleground of non-cancerous cells, signal molecules and structural components that surround and interact with the tumour. It includes immune cells such as macrophages and T cells, as well as stromal cells like cancer-associated fibroblasts (3). A major barrier to successful tumour immunology based treatment is the immunosuppressive TME which contains cells, signals and conditions that restrain the anti-tumour response. The TME facilitates immune evasion through an enrichment of regulatory T cells and M2 macrophages, which can inhibit effector immune cells and secrete immunosuppressive cytokines, including, IL-10 and TGF-B (4). Upregulation of checkpoint proteins like PD-L1 on tumour cells is another cancer escape mechanism used in the TME (3). Immunotherapies like immune checkpoint inhibitors, and CAR T cell therapy, aim to reinvigorate anti-tumour responses to overcome immunosuppression in the TME. Additionally, immune filtration can be an issue due to the increased extracellular matrix in the TME, leading to ‘cold’ tumours (3). As the TME’s immune evasion and immunosuppression is such a significant obstacle to immunotherapies, there is a need to elucidate the mechanisms of action in the TME.
Cancer's signalling sabotage: How intracellular signalling affects tumour immunology?
At the heart of cancer’s defence against the immune system and consequently immunotherapies, lies cancer’s ability to commandeer intrinsic cell signalling pathways that regulate immune recognition and response. Tumours hijack intrinsic signalling pathways to evade immune attacks. Oncogenic mutations to key signalling pathways such as PI3K/mTOR, RAS/RAF/MAPK, Wnt/beta-catenin, STAT3 and NF-kB play dual roles in promoting tumour growth while also mediating an immunosuppressive effect (5). Hyperactivation or dysregulation of these pathways can lead to a decrease in antigen presentation, less T cell infiltration, induction of inhibitory receptors, recruitment of immunosuppressive cells and interference with immune metabolism (5). By understanding dysregulated signalling pathways in tumours, it provides researchers with more targets to drug, to restore immune recognition of tumours.
An infiltration mission: How do we turn ‘cold’ tumours ‘hot’?
Immunotherapies take aim at tumours by harnessing the immune system’s power, but many malignancies reside in immune blind spots, escaping detection. ‘Cold’ tumours occupy these blind spots, as these tumours lack typical immune cell infiltration and inflammation. These ‘cold’ tumours also tend to have a lower mutational burden and display fewer tumour specific antigens (6). Because of the lack of tumour antigens, T-cells are not primed and activated and therefore, do not infiltrate into the TME. Checkpoint inhibitors work by blocking inhibitory signals on T cells, if no T cells are present due to lack of activation, checkpoint inhibitors have no T cells to target. To tackle these ‘cold’ tumours with modern immunotherapies, we need to turn ‘cold’ tumours ‘hot’. Some potential tactics include combination therapies integrating epigenetic modulators like HDAC inhibitors, which can upregulate MHC class I and II expression on tumour cells, making them better targets for T cells (7). HDAC inhibitors have also been shown to shift tumour-associated macrophages from an M2 immunosuppressive to an M1 inflammatory subtype (8). Traditional oncological treatments, like radiotherapy, can also play a role in facilitating the conversion of cold to hot tumours; for example, after radiotherapy, endothelial cells express molecules like ICAM1 which facilitates the attraction of immune cells (9). Encouraging the infiltration of immune cells into cold tumours, could help tip the balance in favour of the immune system, leading to the eradication of tumours.
What are the promises and pitfalls of immunotherapies?
Immunotherapy emerged as a revolutionary paradigm shift in our approach to treating cancer, promising to harness the power of the immune system to target and destroy tumours. The development of adoptive cell therapies and checkpoint inhibitors has improved survival for patients with advanced cancers, particularly in NSCLC and melanoma (10). However, the success of immunotherapy has been limited to a subset of patients. Response rates to PD-1 inhibitors can be as much lower depending on the cancer type due to patients failing to respond or developing resistance. Treatment resistance can arise in several ways, such as loss of antigen presentation, induction of T-cell dysfunction and immunosuppressive cell recruitment. Understanding and overcoming resistance to immunotherapy is essential to increase the number of patients who respond to treatment. To rescue the promise of tumour immunotherapies, an interdisciplinary call-to-arms has been enacted. Immunologists and Microbiologists have been unravelling the impact of the microbiome on clinical outcomes, as the gut microbiome regulates the immune system (11). Machine learning has been used to predict the immunogenicity of neoantigens to develop personalised vaccines (12). Clinicians have been combining checkpoint inhibitors with other therapies like HDAC inhibitors, to improve the efficacy of immunotherapy. Gaining a better understanding of resistance mechanisms in unresponsive patients, is critical to rationally designing new approaches to immunotherapy to defeat cancer.
Conclusion
The struggle between the immune system and cancer rages on, shaping the balance between tumour progression and destruction. To better understand and elucidate these interactions, we present a curated collection of antibodies directed at key topics in tumour immunology.
- Antibodies for immunophenotyping, immune checkpoints, the TME, inflammation, epigenetic changes, and tumour intrinsic signalling pathways.
- Antibodies directed at a range of innate and adaptive immune cells to assist in immunophenotyping cells.
- Antibodies directed at both key co-inhibitory and co-stimulatory molecules in immune checkpoint control to study immune checkpoints.
- Antibodies directed at matrix metalloproteinases, chemokines and hypoxia factors to help understand the interactions in the TME
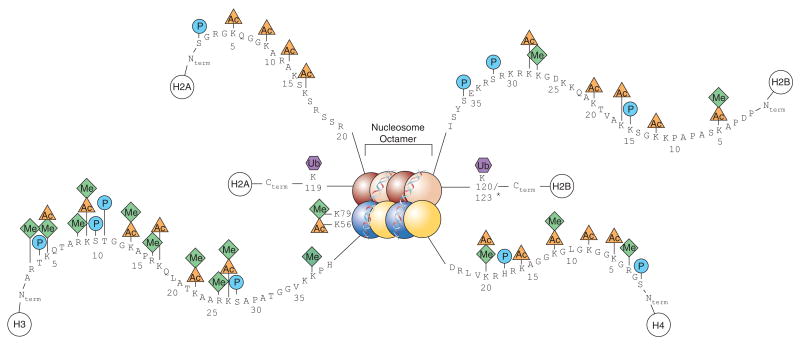
- Antibodies directed at epigenetic changes, and key molecules involved in signalling pathways like MYC and p53.
By supplying these tools, we hope to equip scientists with the antibodies they need to gain deeper insights into how cancer evades and suppresses the immune system, and potentially uncover therapeutic targets.