Written by Dr. Anne E Lykkesfeldt, Danish Cancer Society, Denmark. Inventor of MCF-7 and T47D derivative resistant breast cancer cell lines

Why resistance matters
Resistance to treatment remains a major problem for cancer patients, and understanding the mechanisms which render cancer cells resistant can enable the development of new targeted treatments. Cell culture models are valuable tools for studying these resistance mechanisms and testing new treatments.
In order to gain meaningful insight into these resistance mechanisms, we considered it important to have access to a panel of resistant cell lines representing the diversity seen in patient tumours. In the late 1980s and early 1990s, only few resistant cell lines were available. Therefore, colleagues and I decided to establish resistant cell lines ourselves.
Starting with breast cancer
We were particularly interested in breast cancer, which is the most common cancer disease among women in the western world. The majority of breast tumours are oestrogen receptor (ER) positive. The ER is a transcription factor which requires activation by binding to oestrogenic compounds. Targeted treatment for oestrogen-stimulated tumour growth aims to inhibit the growth-promoting effects of ER. This can be achieved through several approaches including the use of:
- Tamoxifen, a selective oestrogen receptor modulator (SERM) which competes with oestrogen upon binding to the ER and inhibits oestrogen-mediated growth stimulation.
- Fulvestrant, an ER down modulator that binds to the ER and destabilizes the receptor protein, thereby inhibiting oestrogen-stimulated growth.
- Aromatase inhibitors, which block the supply of oestrogenic compounds through the inhibition of both local and systemic oestrogen synthesis by the aromatase enzyme.
The human breast cancer cell lines MCF-7 and T47D have been used as parental cell lines for our MCF-7 and T47D resistance models. Both cell lines derive from patients with metastatic disease.1,2 MCF-7 cells express high levels of ER and normal levels of HER2, and ER is the major driver of growth of MCF-7 cells, whereas HER receptor signalling is not important.3,4 T47D cells have a lower ER level5, normal HER2 level and growth is dependent on both ER and HER receptor signaling.6 We have developed MCF-7 and T47D resistant cell lines against the anti-oestrogens’ tamoxifen and fulvestrant, and we established the MCF-7-derived aromatase inhibitor resistant cell lines against the clinically relevant aromatase inhibitors letrozole, anastrozole and exemestane.
Building resistant cell lines from scratch
Our resistant cell lines were established through clonal selection of cell colonies that survived long term treatment with antihormone concentrations corresponding to the blood levels observed in treated patients (Figure 1).
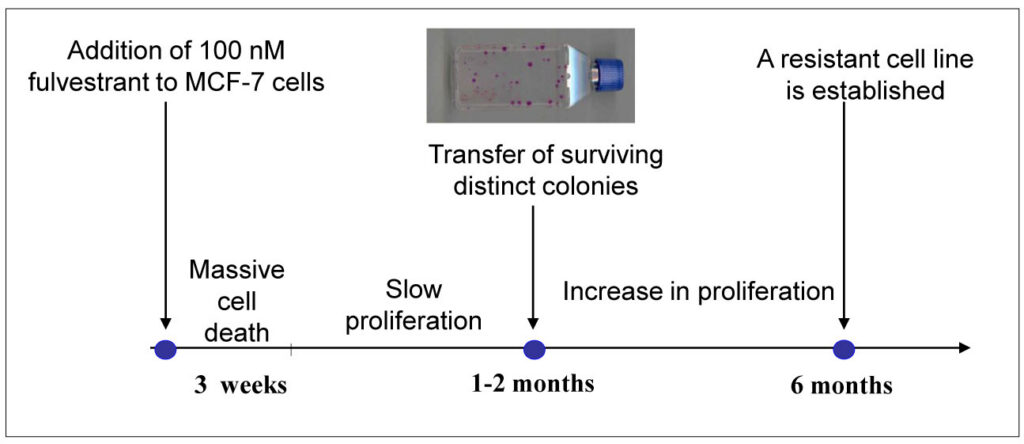
Initially, we observed growth inhibition and cell death but after several weeks, surviving colonies could be isolated and propagated. Gradually, the growth rate increased until they reached a level close to the parental cell line. On average, it took up to 6 months to establish a resistant cell line, which could be passaged once a week. We believe that our model system presents acquired resistance, corresponding to patients who initially respond to treatment and then later relapse. Importantly, all our anti-oestrogen resistant cell lines displayed reduced ER expression and the fulvestrant resistant T47D cell lines became ER-negative. The reduced ER level may play an important role in surviving the long-term anti-oestrogen treatment.
Why a large panel matters
Our large panel of resistant cell lines allows researchers to investigate heterogeneity among cell lines derived from individual clones. It also enables exploration of both differences and similarities in resistance mechanisms associated with specific treatment approaches.
Understanding these resistance mechanisms provides a rationale for selecting new treatments targeting the growth promoting pathways in the resistant cells. New biomarkers may also emerge and support patient-specific treatment selection for personalised medicine.
What we learned about resistance mechanisms
Gene expression analyses of our anti-oestrogen resistant cell lines have shown differential expression of a large number of genes compared with the parental MCF-7/S0.5 cell line.7 Unsupervised cluster analysis and digital gene expression profiling separated the four MCF-7 derived tamoxifen-resistant cell lines from each other and clearly distinguished them from their parental line.8 Several of the differentially expressed genes were oestrogen-regulated genes.7
Significant upregulation of CDK6 expression was found in fulvestrant-resistant cell lines, but not in the tamoxifen resistant MCF-7 models, supporting the notion that distinct mechanisms drive tamoxifen and fulvestrant resistance.9
Kinase screening revealed new targets
Following kinase inhibitor screening of our tamoxifen- and fulvestrant resistant MCF-710 and T47D11 cell lines, we uncovered important biological differences in line with previous findings.
Firstly, a preferential and severe inhibition of the four tamoxifen resistant MCF-7 cell lines was observed with the inhibitor JNJ-770662 targeting Aurora Kinase A/B and CDK1/2 , whereas both the tamoxifen and the fulvestrant resistant MCF-7 cell lines responded to kinases targeting HER-receptor signalling (see Table 1 in ref.10). Aurora kinases A and B are important regulators of mitosis and we found that knockdown of Aurora kinase A resulted in preferential growth inhibition of tamoxifen resistant MCF-7 cells (see Figure 6b in ref.10) and regain of sensitivity to tamoxifen treatment (see Figure 6d in ref.10). This suggests that Aurora Kinase A may act via ER e.g. resulting in the agonistic effect of tamoxifen in tamoxifen resistant breast cancer cells. Regain of sensitivity to tamoxifen treatment in tamoxifen resistant MCF-7 cell lines was also obtained by the multi-targeting kinase inhibitors sorafenib and nilotinib, which exert growth inhibition via ER and ER associated proteins.12
We have previously shown that ER is a major driver of growth in our tamoxifen resistant MCF-7 cell lines. The importance of ER for growth of tamoxifen resistant cell lines is also supported by their sensitivity to fulvestrant treatment.3,15 In fact, our tamoxifen resistant cell line AL-1 (MCF7/TAMR-1) was an early tool demonstrating that tamoxifen-resistant breast cancer cells could be growth-arrested through treatment with a pure anti-oestrogen.14-16
Finally, a kinase inhibitor screen revealed that HER receptor signalling was important for the growth of fulvestrant resistant MCF-7 cell lines.10 This supports our previous finding of increased expression of activated EGFR, HER-3 and Erk-1 in these resistant cell lines.18 As mentioned earlier, all anti-oestrogen resistant cell lines have maintained ER expression, and complete growth arrest of fulvestrant resistant MCF-7 cell lines was obtained by combined treatment against both HER-receptor and ER signalling.4
Differences in T47D resistant models
The T47D cell line is both ER and HER2 receptor positive. Tamoxifen-resistant T47D cell lines showed reduced ER level18 while the fulvestrant resistant variants became ER negative.6 Both tamoxifen and fulvestrant resistant T47D models showed increased c-Src activation11, and treatment with the c-Src inhibitor dasatinib inhibited growth, phosphorylation of c-Src, and signalling via Akt and Erk pathways.6,11
For tamoxifen resistant T47D cell lines, combined treatment against c-Src and ER proved more effective than targeting c-Src alone.11 Further kinase inhibitor screening identified Aurora kinase B as an important driver for the growth of fulvestrant resistant T47D cell lines, and that the Aurora kinase B inhibitor barasertib may be a new treatment candidate for patients with Aurora kinase B positive tumours.19
Looking ahead
Today, treatment with an aromatase inhibitor is first line adjuvant endocrine treatment for postmenopausal women with ER positive tumours. We have MCF-7 derived resistant cell lines for the three clinically relevant inhibitors; letrozole, anastrozole and exemestane19. Eleven of the twelve aromatase inhibitor resistant cell lines were ER positive and the expression level was similar to parental MCF-7 cells. Fulvestrant but not tamoxifen exerted complete growth arrest of the aromatase inhibitor resistant cell lines19. Kinase inhibitor screening revealed Aurora kinase A as an important driver for resistant growth.20 Total growth arrest was achieved through combined treatment with fulvestrant and an inhibitor of Aurora Kinase A.20
Gene expression analysis of tumours from 19 ER positive early-stage breast cancer patients treated with aromatase inhibitor identified 26 genes exhibiting significantly altered expression in patients who showed disease recurrence compared to non-recurrent patients.22 The TFF3 gene encoding the trefoil factor 3, which is an oestrogen responsive oncogene shown to be involved in tamoxifen resistance23 was overexpressed (2.6 fold) in primary cancers of recurrent patients. Higher expression of TFF3 (1.5 fold) was found in our aromatase inhibitor resistant MCF-7/ExeR-1 cell line. Knock-down of TFF3 in MCF-7/ExeR-1 cells significantly reduced cell growth and overexpression of TFF3 in MCF-7 cells abrogated the aromatase inhibitor-mediated suppression of testosterone stimulation, indicating that TFF3 mediates growth and survival signals that overcome the growth inhibitory effect of exemestane.22 Further studies are required to disclose the importance of the 26 identified genes, and our large panel of aromatase inhibitor resistant cell lines is instrumental for these studies, as well as for other studies exploring the mechanisms for aromatase inhibitor resistance.24
MCF-7 and T47D cells are useful for testing new selective ER modulators and orally available pure antioestrogens. Our resistant cell lines are important tools to uncover resistance mechanisms, to find new treatments targets and to find biomarkers to select patients for personalized medicine. The cell lines may also be used for testing new drugs including antibody conjugates and bispecific antibodies. Our models may also be used in co-culture systems with CAR-T cells to explore future therapeutic opportunities.
Conclusion
The resistant cell lines have played a major role for our research and resulted in collaboration and joined publications with many laboratories both in Denmark and abroad.7, 8, 13, 14, 17, 22, 25 In order to test the clinical relevance of the results from our in vitro models, collaboration with Danish Breast Cancer Co-operative Group (DBCG) and European Organisation for Research and Treatment of Cancer (EORTC) has been invaluable and resulted in important discoveries.26-28
Explore the MCF-7 and T47D derivative resistant breast cancer cell lines below.
References
- Soule HD, et al., 1973. Natl Cancer Inst. 51(5):1409-16. PMID: 4357757
- Keydar I, et al., 1979. Eur J Cancer. 15(5):659-70. PMID: 228940
- Thrane S, et al., 2013. Breast Cancer Res Treat. 139(1):71-80. PMID: 23609470
- Sonne-Hansen K, et al., 2010. Breast Cancer Res Treat. 121(3):601-13. PMID: 19697122
- Lykkesfeldt AE, et al., 1986. Br J Cancer. 53(1):29-35. PMID: 3947513
- Kirkegaard T, et al., 2014. Cancer Lett. 344(1):90-100. PMID: 24513268
- Elias D, et al., 2015. Oncogene. 34(15):1919-27. PMID: 24882577
- Lin X, et al., 2013. Breast Cancer Res. 15(6):R1,19. PMID: 24355041
- Alves CL, et al., 2016. CancerClin Cancer Res. 22(22):5514-5526. PMID: 27252418
- Thrane S, et al., 2015. Oncogene. 34(32):4199-210. PMID: 25362855
- Larsen SL, at al., 2015. PloS One.10(2). PMID: 25706943
- Pedersen AM et al., 2014. Int J Oncol. 45 (5): 2167-75. PMID: 25175082
- L R Wiseman, et al., 1993. Eur J Cancer. 29A(16):2256-64. PMID: 8110496
- S Cutrupi, et al., 2012. Oncogene. 31(40):4353-61 PMID: 22249258
- Lykkesfeldt AE, et al., 1994. Cancer Res. 54(6):1587-95. PMID: 8137264
- Lykkesfeldt AE, et al., 1992. Acta Oncol. 31(2):131-8. PMID: 1622627
- Wakeling AE. 1993. Breast Cancer Res Treat; 25:1-9. PMID: 8518404
- Frogne T, at al., 2009. Cancer Res Treat. 114(2):263-75. PMID: 18409071
- Larsen SL, et al., 2015. BMC Cancer. 15:239. PMID: 25885472
- Hole S, et al., 2015. Int J Oncol. 46(4):1481-90. PMID: 25625755
- Hole S, et al., 2015. Breast Cancer Res Treat. 149(3):715-26. PMID: 25667100
- Thomsen KG, et al., 2015. Breast Cancer Res Treat. 154(3):483-94. PMID: 26585578
- Kannan N, et al., 2010. Neoplasia. 12(12):1041-53. PMID: 21170268
- Al-Qasem AJ, et al., 2022. NPJ Precis Oncol. 6(1):68. PMID: 36153348
- Pancholi S, et al., 2008. Endocrine-Related Cancer. 15(4):985-1002. PMID: 18824559
- Rasmussen BB, et al., 2008. Lancet Oncol. 9(1):23-28. PMID: 18083065
- Lykkesfeldt AE, et al., 2009. BMC Cancer. 9:185. PMID: 19531212
- L H Weischenfeldt K, et al., 2017. Acta Oncol. 56(9):1161-1167. PMID: 28488912
